
Меню сайта
Наши новости
Распространение алкалоидов в растительном мире.
Умягчение воды
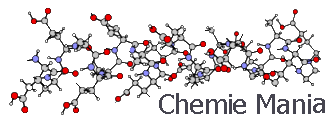
Механическая модель молекулыБиблиотека / Мутации структуры белковоподобного сополимера. Компьютерное моделирование / Библиотека / Мутации структуры белковоподобного сополимера. Компьютерное моделирование / Механическая модель молекулы Механическая модель молекулыСтраница 1
Под моделью системы понимают выбор правил, описывающих взаимодействие частиц между собой и/или с внешними полями, то есть в формулировке вида и способа вычисления функции потенциальной энергии.[8]
В ряде задач физической химии удобно рассматривать молекулу не как электронно-ядерную систему, а как систему взаимодействующих атомов (механическая модель молекулы).
Механическая модель не противоречит квантовой механике, а при некоторых ограничениях (среди них, прежде всего, следует упомянуть теорему Борна-Оппергеймера) может быть выведена из неё.
![]()
![]()
В приближении МО ЛКАО ССП энергия молекулярной системы выражается как сумма одно-центровых взаимодействий (ea), двуцентровых взаимодействий (eab), трёхцентровых взаимодействий ( eabg) и т.д.
(2.1)
Одно-центровые и двуцентровые интегралы естественно отнести к парам ядер (атомов), трёхцентровые интегралы- к тройкам ядер (атомов) и т. д .Тогда получаем энергию молекулы в атом-атомном приближении здесь суммирование ведётся по всем парам атомов, по всем тройкам атомов и т.д.
(2.2)
![]()
![]()
Многочастичные взаимодействия вносят малый вклад в энергию системы, и ими часто пренебрегают.
Парные взаимодействия удобно разбить на взаимодействия атомов, валентно связанных между собой, и взаимодействия атомов, валентно между собой не связанных.
Предполагается, что энергию взаимодействия можно разделить на ряд составляющих.
1. Энергия деформации связи.
При отклонении длин связей от их нормального значения возникает энергия деформация связи (Есв). Обычно связь рассматривают как гармонический осциллятор. При этом можно предположить, что при малых деформациях энергия ковалентных связей подчиняется следующей зависимости.
U(R) = K´(R-Ri)2 (2.3)
Где К – силовая постоянная,
R – равновесная длина связи
Ri – мгновенная длина связи
Рассмотрим вопрос о способе описания валентных связей. Здесь существует альтернатива[9]: 1) моделировать связи каким-либо гладким потенциалом (например гармоническим или потенциалом Морзе), 2) учитывать их как абсолютно жёсткие (геометрические) ограничения. Оба способа имеют свои преимущества и недостатки. В первом случае справедливы простые уравнения Ньютона. Однако из-за высокой "жёсткости" обычных валентных потенциалов связанные частицы могут совершать быстрые колебательные движения, учесть которые можно при очень малом шаге интегрирования. Поэтому даже после весьма большого числа шагов эволюции системы будет на самом деле прослежена лишь в течении короткого интервала времени. Проблема решается путём удаления соответствующих степеней свободы при рассмотрении химических связей как постоянных геометрических ограничений. При этом в системе действует единственный невалентный потенциал, который и задаёт минимальный временной масштаб. Однако в подобной постановке движение частиц описывается уже не уравнениями Ньютона, а уравнениями Лагранжа, что усложняет вычислительную схему и делает её более трудоёмкой. Кроме того, абсолютно жёсткие связи не вполне правильно отвечают реальной ситуации.
Смотрите также
Особенности синтеза и производства витаминов
Производство
витаминов в нашей стране организовано в начале 30-х гг прошлого века. Вначале
выпускались витаминные препараты из натурального сырья. Затем было освоено
производство синтетичес ...
Нанотехнология. Перспективы развития
...
Нестероидные противовоспалительные препараты. Салицилаты
...