
Меню сайта
Наши новости
Распространение алкалоидов в растительном мире.
Умягчение воды
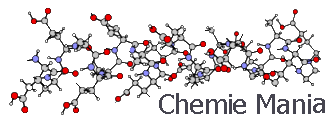
Молекулярное моделирование с помощью программы HyperChemДипломы, курсовые и прочее / Создание новых лекарственных веществ / Дипломы, курсовые и прочее / Создание новых лекарственных веществ / Молекулярное моделирование с помощью программы HyperChem Молекулярное моделирование с помощью программы HyperChemСтраница 1
Молекулярное моделирование – сложная сеть различных наук, находящее применение в нанотехнологии, в молекулярной биологии, квантовой химии и биотехнологии.
Молекулярное моделирование молодая, востребованная и бурно развивающаяся наука.
На сегодняшний день методы квантовой химии и молекулярной динамики получили широкое распространение в численном моделировании электронной и атомной структур сложных систем молекулярных, кристаллических и переходных размеров. Это связано с технологическим развитием соответствующего математического обеспечения. Сейчас в мире функционирует достаточно много современных вычислительных комплексов, реализующих методы квантовой химии и молекулярной динамики. Использование многих из этих методов обеспечивается программой Hyper Chem для молекулярного моделирования.
HyperChem - комплексный программный продукт, предназначенный для задач молекулярного моделирования. Он включает в себя программы, реализующие методы молекулярной механики, квантовой химии и молекулярной динамики. Силовые поля, которые могут использоваться в HyperChem - это ММ+ (на базе ММ2), Amber, OPLS и BIO+ (на базе CHARMM). Реализованы полуэмпирические методы: расширенный метод Хюккеля, CNDO, INDO, MINDO/3, MNDO, AM1, PM3, ZINDO/1, ZINDO/S, а также возможности проведения неэмпирических расчетов и по теории возмущений Меллера-Плессета второго порядка.
HyperChem обладает развитыми средствами визуализации, которые могут использоваться как при подготовке входной информации (структуры молекулы), так и при анализе результатов, например, рассчитанных характеристик ИК- и УФ- спектров.
Расчётные методы оказывают неоценимую помощь в создании лекарственных средств. Молекулярное моделирование входит во все области знаний и находит себе применение, порой играя одну из главных ролей. Некоторые области химии немыслимы без молекулярного моделирования. В развитых странах моделирование является современным методом изучения микроструктур.
В настоящее время для изучения реакционной способности молекул используются приближения CNDO/2, MNDO, AM1, PM3.
Метод CNDO основан на приближении нулевого дифференциального перекрывания и поэтому является одним из простейших полуэмпирических методов. Из этого факта следуют ограничения применимости метода, который из-за обедненной расчетной схемы недостаточно корректно воспроизводит многие эффекты. С появлением более совершенных версий полуэмпирических методов МО приближение CNDO все реже применяется на практике. Так, в версии 7 программного продукта МОРАС данный метод не представлен. Тем не менее, во многих случаях для быстрой оценки электронных параметров полезно использовать схему CNDO, так как вследствие резкого уменьшения количества рассчитываемых интегралов с помощью этого метода можно исследовать более сложные объекты. В целом CNDO/2 дает надежные результаты при расчете электронных распределений и свойств, зависящих от них.
Основным калибровочным параметром в CNDO является резонансный интеграл. Он подбирается так, чтобы относительный порядок энергетических уровней занятых МО и коэффициенты разложения МО в ЛKAO наилучшим образом совпали с расчетами ab initio соединений обучающей выборки.
Общим достоинством всех перечисленных версий является прежде всего сравнительно малое время расчетов и меньшие размеры занимаемой оперативной памяти по сравнению с более точными приближениями. Это дает возможность как для быстрой оценки исследуемых объектов, так и для изучения более сложных молекул, требующих длительного времени расчета и больших объемов оперативной памяти. В целом приближение CNDO хорошо описывает электростатические эффекты и полярность связи. CNDO/2 может применяться для расчета дипольных моментов и зарядов по схеме Малликена и оценки равновесной геометрии.
Недостатки приближения CNDO являются следствием усечения расчетной схемы, которая не учитывает взаимодействия между перекрываниями орбитальных зарядов. В результате многие эффекты не воспроизводятся.
Метод MNDO был разработан на основе более строгого и сложного приближения NDDO. Это позволило существенно улучшить результаты расчетов при решении многих задач. Длительное время метод рассматривался в качестве основного полуэмпирического метода квантовой химии. Его возможности позволили с достаточной степенью надежности рассчитывать физико-химические свойства, электронные структуры и реакционную способность множества молекулярных систем.
Преимущество заключается в быстродействии (по сравнению с неэмпирическими методами) программ, в которых реализована схема MNDO. Это позволяет применять ее для исследования все более сложных объектов. Недостатки связаны с тем, что точность метода не может превышать точность тех экспериментальных данных, по которым проводилась параметризация.
Страницы: 1 2
Смотрите также
Проблемы и решения на уровне учения о химических процессах
Учение о
химических процессах - это область науки, в которой существует наиболее
глубокое взаимопроникновение физики, химии и биологии. На этом уровне развития
химических знаний химия становится на ...
Азотная кислота
...
Исследование морфологической структуры, физико-химических и химических характеристик беленой, сульфатной целлюлозы из древесины хвойной породы
Одним
из наиболее важных факторов, определяющих развитие большинства отраслей
промышленности, является устойчивая сырьевая база, и в частности углерод
содержащее сырье. К такому сырью относ ...