
Меню сайта
Наши новости
Распространение алкалоидов в растительном мире.
Умягчение воды
Развитие методов расчета молекулярных орбиталейОрганическая химия / Атомные и молекулярные орбитали / Органическая химия / Атомные и молекулярные орбитали / Развитие методов расчета молекулярных орбиталей Развитие методов расчета молекулярных орбиталейСтраница 2
Однако даже при хорошем, правильно выбранном базисе энергии молекул, рассчитанные методом ССП (относительно энергий ядер и электронов - обычной точки отсчета в квантово-химических расчетах), всегда больше экспериментальных значений. Это связано с тем, что учитывается только усредненное во времени электростатическое взаимодействие между электронами. На самом деле движение любого электрона коррелирует с движением любого другого электрона, находящегося в его окрестностях. Если на орбиталях около одного ядра находятся два электрона, и один из них расположен вблизи ядра, то второй электрон имеет меньшую вероятность находиться вблизи ядра по сравнению с вероятностью в отсутствие первого электрона. Такая корреляция движения электронов (электронная корреляцияч) уменьшает электростатическое отталкивание между электронами и стабилизирует молекулу.
Для учета энергии электронной корреляции чаще всего применяют метод конфигурационного взаимодействия. На орбиталях, полученных методом Хартри-Фока, электроны можно разместить по-разному. Определенное размещение электронов по орбиталям называется электронной конфигурацией. Основному состоянию молекулы соответствует электронная конфигурация ψ0. В обычных молекулах в электронной конфигурации ψ0 все электроны расположены парами на низших орбиталях. При возбуждении одного электрона возникает однократно возбужденная конфигурация, при возбуждении двух электронов - двукратно возбужденная конфигурация и т.д. Линейная комбинация невозбужденной и различных возбужденных электронных конфигураций дает полную волновую функцию молекулы, в которой уже учтена энергия электронной корреляции. При учете всех возможных конфигураций (в рамках данного базисного набора) можно прийти к пределу полного конфигурационного взаимодействия.
Ниже приведена карта Хегре-Радома-Шлайера-Попла, на которой показаны пути улучшения расчетов молекул. Работа химика-теоретика проводится в рамках центрального поля, которое на карте заштриховано. Простейшая модель молекулярных орбиталей получается методом Хартри-Фока с минимальным базисом Улучшение базисного набора соответствует движению по карте вниз. Движение слева направо соответствует улучшению способа учета электронной корреляции. Исследования ведутся по пути улучшения или базисного набора, или электронной корреляции, или того и другого.
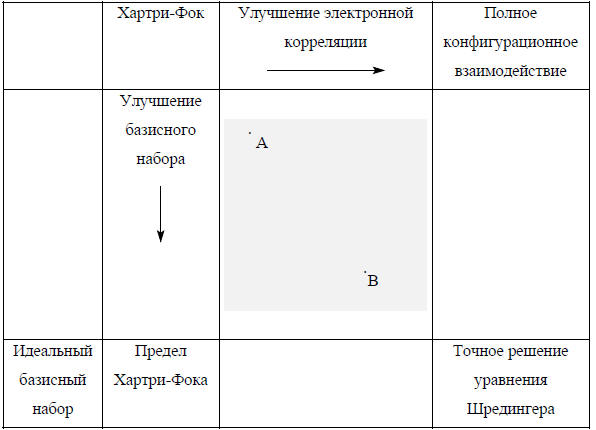
Метод, указанный на диаграмме точкой В, находится на более высоком теоретическом уровне, чем метод А, поскольку точка В по сравнению с точкой А лежит правее и ниже. Однако метод на более высоком теоретическом уровне не обязательно даст более точные результаты расчета конкретных свойств молекул. Часто более точные методы получают методом более низкого теоретического уровня (А), чем методом более высокого уровня (В). В каждом конкретном случае в соответствии с имеющейся документацией выбирают тот метод расчета, который наиболее адекватен поставленной задаче.
В настоящее время широкой известностью пользуется метод, основанный на математической теории функционала плотности (DFT). В рамках этого метода, например, удалось теоретически объяснить жесткость и мягкость кислот и оснований (глава 3). Метод DFT скорее следует отнести к полуэмпирическим, чем к неэмпирическим, поскольку для достижения наилучших результатов часто необходимо использовать не один, а несколько функционалов.
Страницы: 1 2
Смотрите также
Фазовые равновесия в системе MgS-Y2S3
Соединения с участием
РЗЭ остаются по прежнему обширным резервом для создания новых материалов.
Возможно создание материалов с уникальными, заранее заданными свойствами.
Взаимодействие в
...
Введение.
В настоящее
время в резиновой промышленности применяют широкий спектр каучуков, однако
большую часть промышленного потребления составляют натуральный и синтетический
полиизопрены. До сих пор натура ...